Clinical Research
Of NIH’s more than $18B investment in clinical research, clinical trials (~$7B) reflect the point at which the public is most directly engaged in NIH’s clinical research activities, either as dedicated volunteer research participants or users of the resulting data and health interventions. Successful management and oversight of the clinical trial enterprise remains essential to NIH’s mission to translate basic biomedical discoveries into improved health outcomes. OSP works to ensure that NIH’s clinical trial policies enhance the design, conduct and oversight of clinical trials.
Clinical Trials
NIH Definition of Clinical Trial
A research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes.
- Notice of Revised NIH Definition of “Clinical Trial” Revised NIH Clinical Trial Definition
- NIH’s Clinical Trial Definition
- Does Your Human Subjects Research Study Meet the NIH Definition of a Clinical Trial?
Sharing Clinical Trial Information
HHS Final Rule for Clinical Trials Registration and Results Information Submission
- Federal Register Notice of Final Rule for Clinical Trials Registration and Results Submission
- NPRM Clinical Trials Registration and Results Submission (November 2014)
- Summary of the Final Rule and NIH Policy
- Key Elements of Final Rule and NIH Policy
- Summary Table of Changes from Current Practice Described in the Final Rule
- Federal Register Notice for NIH Policy on the Dissemination of NIH-Funded Clinical Trials
- NIH Guide Notice
GCP Training for NIH Awardees Involved in NIH-funded Clinical Trials
Clinical Trial Enrollment Workshop (July 2015)
NIH Workshop on the Enrollment and Retention of Participants in NIH-funded Clinical Trials July 25, 2014
Presentations:
Resources
- ClinicalTrials.gov
- Clinical Trial Requirements for Grants and Contracts
- ICH E6 (R2) Good Clinical Practice (GCP) Integrated Addendum to ICH E6 (R1)
- New Review Criteria for Research Project Applications Involving Clinical Trials
- NIH ClinRegs
- NIH Clinical Trials and You
- NIH Stem Cell Clinical Trials & Other Information
- Regulations: Good Clinical Practice and Clinical Trials
- Research Methods Resources
- OSP-OER Blog “Improving Visibility of NIH-Supported Clinical Trial Activities and Results Information”
- OSP-OER Blog “Building Better Clinical Trials through Enhanced Stewardship and Transparency”
- JAMA Article “Toward a New ERA of Trust and Transparency in Clinical Trials”
- NEJM Article “The Final Rule of U.S Clinical Trials Registration and Results Information Submission”
Clinical Trial E-Protocol Tool
Clinical Trial E-Protocol Tool and Template Documents
The electronic protocol writing tool aims to facilitate the development of two types of clinical trials involving human participants. The first type of trials are Phase 2 and 3 clinical trial protocols that require a Food and Drug Administration (FDA) Investigational New Drug (IND) or Investigational Device Exemption (IDE) application.
NIH developed a second protocol template to help behavioral and social science researchers prepare research protocols for human studies measuring a social or behavioral outcome or testing a behavioral or social science-based intervention.
Both templates found in the electronic protocol tool meet the standards outlined in the International Council on Harmonisation (ICH) Guidance for Industry, E6 Good Clinical Practice: Consolidated Guidance (ICH-E6). These are international standards of good clinical practice that apply to all clinical trials, and their goals are to ensure research integrity and protect human subjects. In addition, use of the electronic protocol tool allows researchers to interface directly with clincialtrials.gov.
Take me to the Web-based e-Protocol Writing Tool
- Final “Phase 2 and 3 Clinical Trial” Template Documents
- Final “Behavioral and Social Sciences Research Involving Humans Template” Documents
Informed Consent
As a steward of the nation’s biomedical research enterprise, NIH is dedicated to ensuring that data and biospecimens are shared for research ethically, securely, and with respect for the privacy, autonomy, and well-being of research participants and the communities to which they belong. Responsible sharing of data and biospecimens derived from human participants relies on robust informed consent practices that uphold the principles of autonomy and trust in biomedical research. Fundamental to these practices are clear, efficient, and transparent communication strategies for conveying potential risks and benefits of sharing, enabling individuals to make informed decisions to participate in research, retain autonomy in decision making, and understand potential uses and contributions of their data and specimens.
OSP works to ensure robust informed consent practices are understood and in place across NIH, with the goal of protecting research participants altruistically donating data and specimens to advance the scientific enterprise.
Resources
Informed Consent for Secondary Research with Data and Biospecimens: Points to Consider and Sample Language for Future Use and/or Sharing
- Informed Consent for Secondary Research with Data and Biospecimens: Points to Consider and Sample Language for Future Use and/or Sharing
- Request for Information: Developing Consent Language for Future Use of Data and Biospecimens
- Public Comments
Informed Consent for Research Using Digital Health Technologies: Points to Consider & Sample Language
NIH Single IRB (sIRB) Policy
The NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research establishes the expectation that all sites participating in multi-site studies involving non-exempt human subjects research funded by the National Institutes of Health (NIH) will use a single Institutional Review Board (sIRB) for all U.S. sites to conduct the ethical review required by the Department of Health and Human Services regulations for the Protection of Human Subjects. If you have any questions about this policy, please contact us here.
Relevant Documents and Resources
- Final NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research
- Federal Register Notice on the Final NIH sIRB policy
- Federal Register Notice on sIRB Effective Date Extension
- NIH Guide Notice on the Final sIRB policy
- NIH Guide Notice on sIRB Effective Date Extension
- NIH Director’s Statement on the NIH sIRB policy
- OSP-OER Blog on the sIRB policy
- NCATS SMART IRB Reliance Platform
- Frequently Asked Questions about the Implementation of the sIRB policy
- NIH Guide Notice on Scenarios Illustrating the Use of Direct and Indirect Costs for Single IRB Review Under the sIRB Policy
- NIH Policy on the Use of a Single IRB for Multi-Site Research FAQs on Costs
- Request for Comments on the Draft NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research
- Public Comments on the Draft Policy
- New Federal Register Notice regarding the extension of the effective date for the Single IRB policy
Single IRB Review for Multi-Site Research Resource and Infrastructure Development Workshop (September 2018)
On September 12, 2018, NIH held a Workshop on Single IRB Resource and Infrastructure Development. The workshop focused on successful strategies and lessons learned for modifying and enhancing institutional IRB infrastructure for single IRB review of multi-site studies.
Agenda
Biographies
Relevant Information and Resources
Presentations:
SESSION I – Customizing eIRB Systems to Review Multisite Studies Using a Single IRB Model, Medical University of South Carolina
SESSION I – Models for Institutions Serving as sIRB of Record, Washington University
SESSION I – Single IRB Standard Best Practices, New York University
SESSION II – Models for Assisting Institution Relying on sIRB – Partnership and Innovation Exploring Single IRB Models to Support Clinical and Translational Research, Yale University
SESSION II – Models for Assisting Institutions Relying on sIRB – Incorporating sIRB Procedures into an Existing Research Network, University of Rochester
SESSION III – Facilitating Single IRB Review for Multi-site Research:The OneIRB IT Platform, University of Penslyvania
SESSION III – Administrative Supplements for CTSA Awardees: Development of Resources to Facilitate Single IRB Review for Multi-Site Research, University of Cincinnati
Webcast
Data and Safety Monitoring
NIH is committed to supporting research studies that are scientifically sound and rigorously designed to produce high quality, reliable scientific data that advances the health of all Americans. NIH provides oversight and stewardship of the clinical research enterprise, supporting the most meritorious and rigorously designed studies, and ensuring our studies adhere to the highest ethical standards.
NIH strives to ensure that all NIH-supported clinical research generates high quality data and holds paramount the safety of all participants. Monitoring is foundational to ensuring scientific rigor, participant safety, accountability and enhancing clinical research efficiency. It is also a key component of NIH’s vision for all clinical research.
Modernizing Data and Safety Monitoring
To enhance efficiency and rigor, NIH intends to modernize the approach to monitoring and is committed to engaging the public in development of an updated policy. Since the 1998 NIH Data and Safety Monitoring (DSM) Policy was published, study designs and monitoring practices have evolved, creating the need for more efficient and flexible practices to monitor data for all clinical research. NIH has a phased plan to modernize the DSM Policy starting with gathering input from the public, DSM experts, and the research community.
NIH’s plan of action for developing a modernized Data and Safety Monitoring Policy is below.
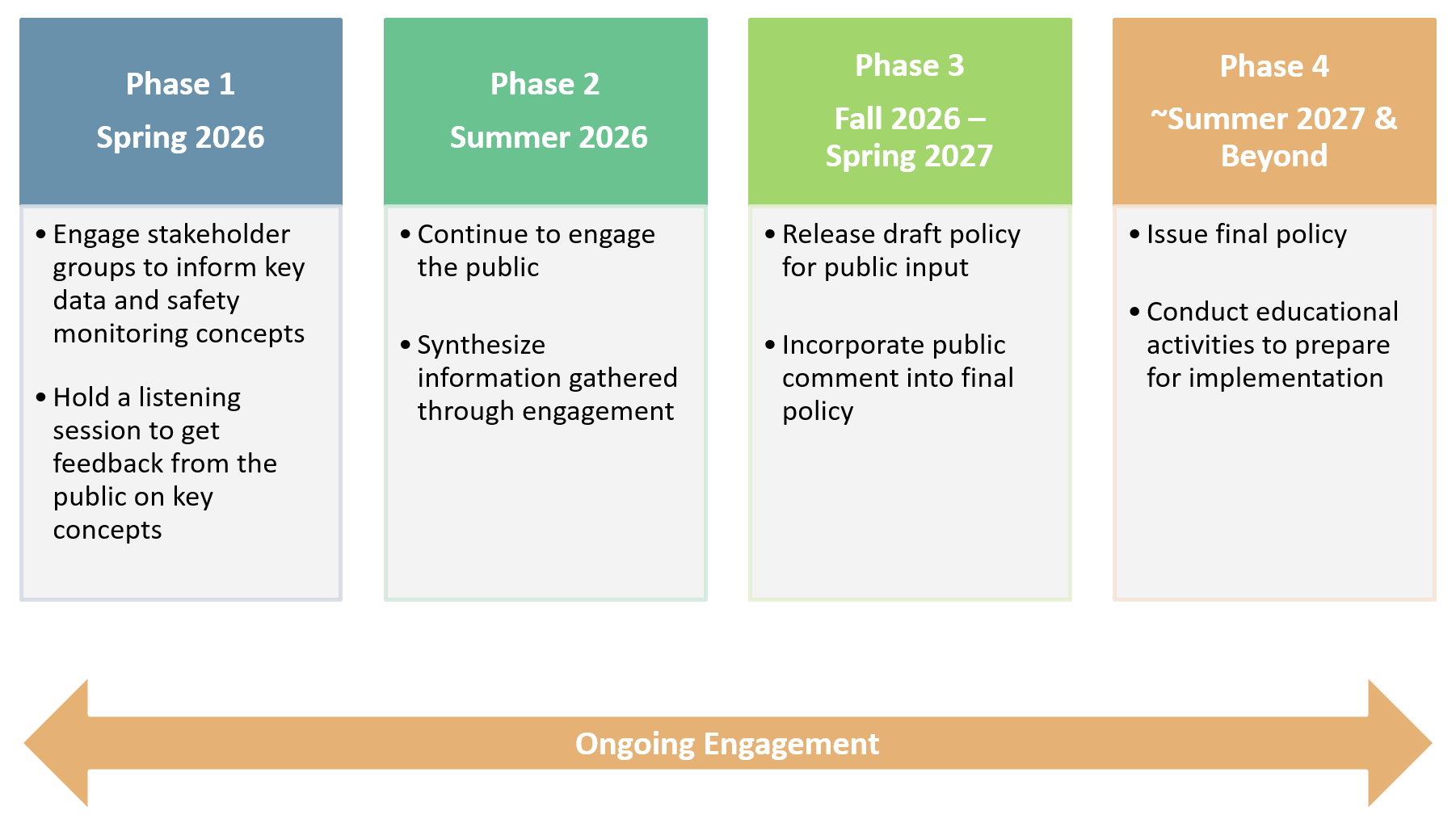
Public Engagement in Developing a Draft Policy
Public engagement is a key component in the successful development of the new Data and Safety Monitoring Policy. We need to hear from a wide array of stakeholders to ensure we develop a responsive, forward-looking, and flexible policy. NIH plans to host a listening session webinar where stakeholders can provide feedback. There is also an opportunity for on-demand commenting where interested parties can provide feedback 24/7 until the draft policy is released. Members of the public will also have the opportunity at that time to provide feedback on the draft policy.
To learn more about on-demand commenting, please click here.
For more information on the webinar, please click the flyer below.
Current NIH Data and Safety Monitoring Policy
In 1998 and 2000, NIH issued the NIH Data and Safety Monitoring (DSM) Policy and Further Guidance on Data and Safety Monitoring for Phase I And Phase II Trials. The DSM policy expects each NIH Institute and Center (IC) to have a system for appropriate oversight and monitoring of clinical trials to ensure the safety of participants and the validity and integrity of the data. In 2020, NIH issued the Data and Safety Monitoring Board Brief to provide clarity on the role of Data and Safety Monitoring Boards. The NIH requirement for data and safety monitoring is separate and distinct from the requirement for protocol review and approval by an Institutional Review Board (IRB).
Documents and Resources
- Office for Human Research Protections (OHRP): Guidance on IRB Continuing Review of Research
- FDA Guidance for Clinical Trial Sponsors: Establishment and Operation of Clinical Trial Data Monitoring Committees
- Draft FDA Guidance on the Use of Data Monitoring Committees in Clinical Trials (February 2024)
- Guideline on Data Monitoring Committees (European Medicines Agency, July 27, 2005)
- Healthcare Research and Quality (AHRQ) Data and Safety Monitoring Policy
Protections for Participants in Research (Human Subjects Research Protections)
Participants in NIH-funded clinical research studies receive a robust set of protections in accordance with standards established by the U.S. Department of Health and Human Services (HHS) in 45 Code of Federal Regulations (CFR) part 46 (“the Common Rule”), and, when applicable, Food and Drug Administration (FDA) Regulations for 21 CFR Part 50 and 56. Additional protections for research participants are also established under Section 2012 and 2013 of the 21st Century Cures Act, the NIH Certificates of Confidentiality policy, the Privacy Act, and the HIPAA Privacy Rule. Protections provided by the Common Rule ensure the safety, rights, and welfare of participants in research (what the Common Rule calls “human subjects”) under a framework that prioritizes the ethical principles enshrined in the Belmont Report (i.e., respect for persons, beneficence, and justice). These protections include oversight of research by Institutional Review Boards (IRBs), as well as requirements for informed consent, confidentiality, and privacy.
OSP serves as a resource for and advisor to the NIH research community with respect to the implementation of regulations, laws, and policies impacting the protection of participants in NIH research. OSP advises on harmonization and optimization, works with NIH Institute, Center, and Office (ICO) partners to identify and resolve policy issues, and recommends and develops new policies as needed. OSP works closely with colleagues across NIH and HHS, including NIH ICOs as well as FDA, the Department of Defense (DOD), the Department of Veteran’s Affairs (VA), the HHS Office for Human Research Protections (OHRP), and the HHS Secretary’s Advisory Committee on Human Research Protections (SACHRP).
Relevant Documents and Resources
Implantable Device Clinical Trials
NIH supports a broad portfolio of clinical trials that implant investigational medical devices (“implant trials”) with the potential for life-changing or life-saving outcomes. Some examples include: the use of spinal cord stimulation to restore movement and bladder function in people with spinal cord injury, development of an ocular implant to treat neurodegenerative retinal disease, and the use of deep brain stimulation (DBS) to mitigate treatment-resistant schizophrenia.
Participants in implant trials have unique post-trial needs that may not be adequately met through existing mechanisms of support. They may face ongoing care needs and potential explantation (removal) surgery. Post-trial care is often complex, and while clinical trials have discrete endpoints, implant trial participants’ needs and interests often extend beyond the study’s conclusion, making it essential to anticipate post-trial care needs during trial planning to allow for continuity of care. OSP and the NIH Office of Extramural Research (OER) coordinate efforts across the agency to consider potential solutions and develop resources to safeguard the welfare of participants enrolled in implant trials.
Request for Information on Draft NIH Resources to Support Implantable Device Trials
On March 25, 2026 NIH released a request for feedback on two draft resources intended to support the research community in planning for the post-trial needs of participants and ensuring transparency during informed consent. More information about these resources can be found at https://grants.nih.gov/grants/guide/notice-files/NOT-OD-26-061.html. Comments on the proposal must be submitted through the comment form found at https://osp.od.nih.gov/comment-form-draft-nih-resources-to-support-implantable-device-trials/. Comments will be accepted until May 25, 2026.
NIH has also created a flyer that can be sent to stakeholders with all the information needed to provide comments. To download, please click here.
Privacy and Confidentiality in Research
Privacy and confidentiality are essential components of a robust research enterprise. NIH takes seriously the obligation to ensure privacy and confidentiality for individuals who have altruistically provided data and samples for research. Some of the obligations for these protections are outlined in the Common Rule, which describes a framework for protecting the privacy and confidentiality of sensitive, private information. Additional protections are provided by Section 2012 and 2013 of the 21st Century Cures Act and the NIH Certificates of Confidentiality Policy, which ensure appropriate protections for research when identifiable, sensitive information is collected or used. The Privacy Act and HIPAA Privacy Rule outline further protections for when individual data subject to these laws can be released.
OSP serves as a resource and advisor to the NIH research community on privacy and confidentiality regulations, laws, and policies. OSP advises NIH ICOs on these protections and works with ICOs to identify and resolve any policy issues.
Relevant Documents and Resources
- Common Rule: 45 CFR Part 46
- 21 CFR part 50 and 56 FDA Regulations for the Protection of Human Subjects
- Section 2012 and 2013 of the 21st Century Cures Act Provision on Certificates of Confidentiality
- NIH Certificates of Confidentiality policy
- NIH Certificates of Confidentiality website
- Privacy Act
- HIPAA Privacy Rule
- 2013 Privacy Rule Amendments
- HIPAA Privacy Rule and Its Impacts on Research (nih.gov)